


Japan Market Access Forum (J-MAF) Updates
2025.6.16
Expanding the Global Reach of Japan-Based Medical Device Start-Ups
On Friday, Astellas Pharma announced that they will allow biotech startups in Japan to access their open-innovation lab and office space in Tsukuba, located just northeast of Tokyo. They will also provide consultation services concerning development – including strategies to expand to markets outside Japan. We think this is a great initiative to help promote drug development in Japan and, hopefully, to help promising Japanese drug developers “Go global” with their innovative treatments. Japan’s healthcare market is one of the largest in the world, but its growth rate will decline as the population continues to decline over the next few decades. So, accessing global markets will be even more critical for Japanese drug manufacturers moving forward.
This is true for Japanese medical device manufacturers as well. While the growth rate of the medical device market in Japan is expected to be higher than that of the drug market over the next decade, Japan’s medical device market, too, is destined to decline. To maintain their current growth rates Japanese device manufactures must also consider expanding their markets outside Japan. Last month Nipro, one of Japan’s largest medical device manufacturers, announced that it aims to increase the share of its business from overseas sales from its current level of 68% (not bad!) to 77% by 2030 and to increase its share of the global dialyzer market from 22.5% to 27.9% - or about 12.6% CAGR. Sysmex is also aiming to expand the overseas sales of its "hinotori™ Surgical Robot System", Japan’s first Made-in-Japan robotic-assisted surgery system, with its first case performed in Malaysia earlier this year.
Moreover, with the COVID pandemic and the technology supply constraints stemming from the Russo-Ukrainian War, some issues with over-reliance on the importation of medical devices from outside Japan have been highlighted. In addition to promoting the development and commercialization of medical devices, the Japanese government’s recently published 2025 Basic Policy on Economic and Fiscal Management and Reform also emphasizes the importance of reinforcing the supply chain for medical devices and pharmaceuticals, and securing domestic production systems. So, maintaining a healthy environment for manufacturing medical devices in Japan is now a national priority.
Although direct assistance from large device manufacturers to medical device start-ups in Japan, like that announced by Astellas for Japan-based pharms start-ups on Friday, may be limited, we recently learned of three local initiatives that aim to support medical device developers in Japan: the Fukushima Medical Device Development Support Centre (FMDSC) started in 2016, the Osaka Chamber of Commerce and Industry Medical Device Industry Forum (MDF) started in 2003, and the Organization to Promote the Healthcare and Medical Device Industry in K(Q)yusu (HAMIQ) established in 2013. There websites are shown below:
Fukushima Medical Device Development Support Centre (FMDSC)
https://fmddsc.jp/en/
Osaka Chamber of Commerce and Industry Medical Device Industry Forum (MDF)
https://www.osaka.cci.or.jp/mdf/consultation/
Organization to Promote the Healthcare and Medical Device Industry in K(Q)yusu (HAMIQ)
https://hamiq.koic.or.jp/
These organization are staffed and supported by seasoned medical device experts in Japan and provide a range of services to Japan-based medical device developers – including testing labs, development consultation, and matching support services in Japan and outside Japan. Some are also supported by industry groups and/or larger medical device manufacturers and distributors. While they report many success cases, it is unclear how much their efforts have contributed to commercialization of novel medical devices outside Japan. However, the range of services that they provide to medical device developers in Japan that are willing to take advantage of their support is impressive.
While not exclusively for Japan, we have also noticed some industry led initiatives to help Japanese companies expand their horizons. The Asia-Pacific MedTech Acuator initiative, for example, provides a good opportunity for some companies to share their technologies regionally. The Medtech Innovator Asia Pacific initiative also recognizes and supports promising new technologies originating from Asia-Pacific countries. There also some country-specific collaborative groups like the US-Japan Healthcare Connection (formerly US-Japan MedTech Frontiers) which aims “to share best practices for medical device innovation and promote networking and collaboration between US and Japanese medical device organizations.” There are also a few other support initiatives that are less specific to the medtech sector, such as the Life Science Innovation Network Japan (LINK-J) and the Ministry of Economy, Trade and Industry’s (METI) Healthcare Innovation Hub.
GinePro primarily supports overseas medical device and regenerative medicine companies with market access and evidence generation for Japan, but we recognize that there is some great innovation taking place in Japan that may also address unmet needs in markets outside Japan [1]. So, a more fluid, two-way collaboration to bring new medical technologies to Japan and to introduce “Japan-borne” medical devices to markets outside Japan may be needed. The key is probably to find products that match the focus and core strengths of each party involved that will lead to a “win-win” for both sides. There may also be a need for more information sharing and education about innovative devices originating from Japan. As the expression goes, "If a tree falls in the woods and no one is around to hear it, does it make a sound?"
GinePro will work to support those kinds of information needs and collaborations moving forward! For example, we will be attending the Japan Health event in Osaka from June 25-27 and we are looking forward to meeting with medical device companies from around the world – as well as some of the country-specific industry groups that are supporting those companies. Drop us a line if you plan to attend!
Footnote:
[1] We were enthralled recently by a presentation by Dr. Yutaka Maruoka, Director of the Department of Oral and Maxillofacial Surgery, National Center for Global Health and Medicine, Japan Institute for Health Security, depicting his journey in developing the Vivomark tissue marker pen together with Mitsubishi Pencil Co., Ltd., and others. The Vivomark tissue marker pen, which is sold by Yasui Co. Ltd. in Japan, allows surgeons to draw precise, thin lines on bones using ink that is safe for the human body. It was born from a real medical need and overwhelming concern about the safety of its predecessor products. You can find information on the Vivomark pen and other innovative products sold by Yasui on their website below: https://yasuico.com/new-medical-devices-surgery/
2025.5.30
Foreign Reference Pricing for New Devices at Initial Listing
President Trump’s Executive Order earlier this month that established Most-Favored Nation (MFN) pricing for the Centers for Medicare and Medicaid Services (CMS), the world’s largest single purchaser of prescription drugs, sparked a lot of discussion among the market access specialists and pharma industry professionals in my network. The question is how drug manufacturers will adjust their planning and launch strategies around the world, given that the US will be referencing the lowest prices from a basket of other developed countries. It will be interesting to see how things play out. Japan references the US, UK, Germany, and France for new drug listings and will make an upward or downward adjustment if the price initially calculated for Japan, based on the calculation rules we use, is less than 75% or more than 125% of the average foreign reference price.
Although medical devices may not be affected by the new Executive Order, foreign reference pricing for medical devices in Japan has been common practice for over two decades. Japan currently references market prices in the US, UK, Germany, France, and Australia and considers the average price for those markets. While foreign reference pricing only happens once in Japan for prescription drugs, for medical devices it is considered both at initial listing and when the actual market price exceeds the average foreign reference price by 1.3 times. There is also a rule for medical device reimbursement in Japan that says that the initial reimbursement price in Japan cannot exceed 1.25 times the average foreign reference price. However, for certain novel devices, those designated as high medical needs devices, or those that receive orphan device designation or Specific use designation, the upper limit of the reimbursement price requested for Japan is 1.5 times the average foreign reference price.
We did a quick review of the ratio of the average foreign reference prices mentioned in listing reports for newly reimbursement prices to the price initially calculated for Japan for unique devices newly reimbursed between fiscal year (FY) 2015 and FY 2024 in Japan. To avoid double counting, we excluded some device components that were clearly part of the same device set and had the same price / price ratio for each component in the set. Also, devices that did not report a reference price for any of the reference markets were excluded – which accounted for about one-third of newly reimbursed devices.
In total we reviewed 137 devices newly listed between FY 2015 and FY 2024. In Figure 1 below, you can see that for nearly 70% of devices, the initial reimbursement price for Japan was less than 100% of the average foreign reference price meaning that the initial price for Japan is typically less than average foreign reference price. However, just looking at the 73 devices that were newly reimbursed between FY 2020 and FY 2024, only about 58% were less than 100% of the average foreign reference price compared to around 80% for those newly reimbursed between FY 2015 and FY 2019. So, while the situation seems to have improved in recent years, the initial reimbursement price is still typically less than the average foreign reference price for newly listed devices in Japan.
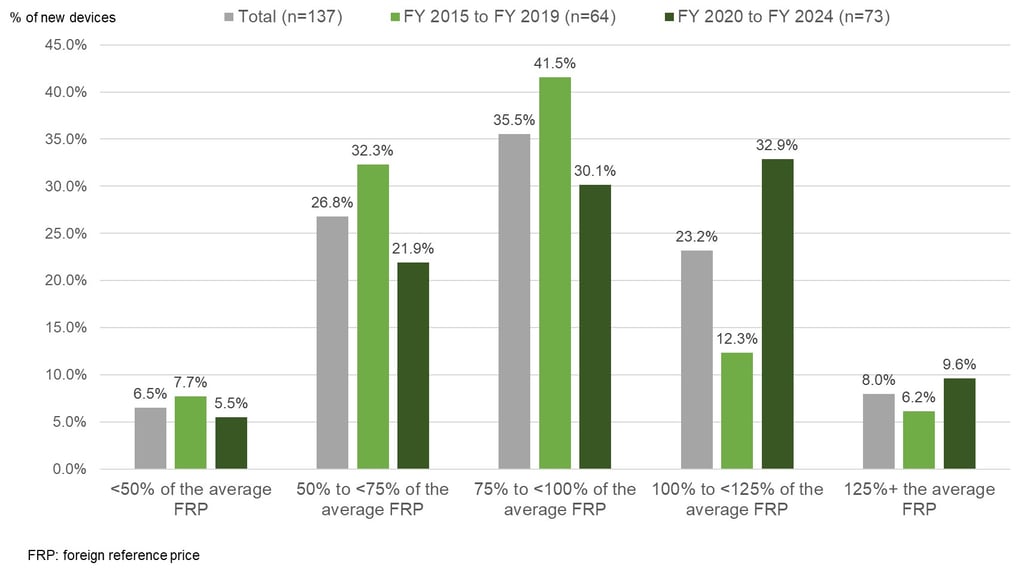
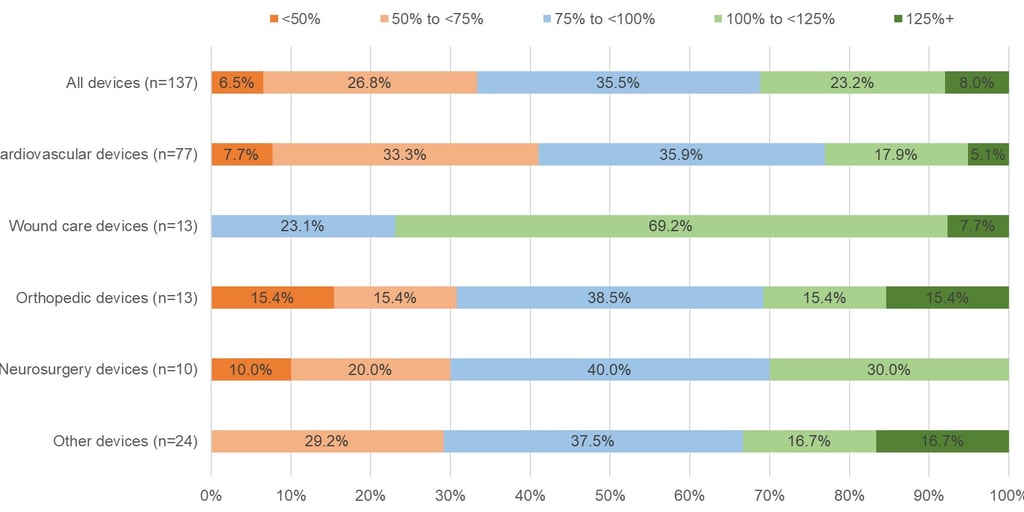
In Figure 2 below you can see how the situation differs by therapy area. The sample sizes are small, but the initial reimbursement price for devices treating cardiovascular conditions in Japan is more often below the average foreign reference price at listing. This is interesting given that a recent study showed that devices treating cardiovascular conditions are more likely to receive a reimbursement premium (see here). Again, while the sample size is small, wound care products seem to have done well, with around 77% of those products receiving an initial reimbursement price in Japan that was at least the same as – if not more than – the average foreign reference price.
Lastly, Figure 3 below shows what the ratio is typically like for the price in each of the reference markets versus the initial price calculated for Japan. Overall, the initial price calculated for Japan is about 85% of the average foreign reference price, on average, but there were large differences by country. The initial reimbursement price for Japan was, on average, about 68% of the US market price, 85% of the UK market price, 89% of the Germany market price, and 99% of the Australia market price. However, the initial reimbursement price for Japan price was often higher than the market price in France at 102%, on average.
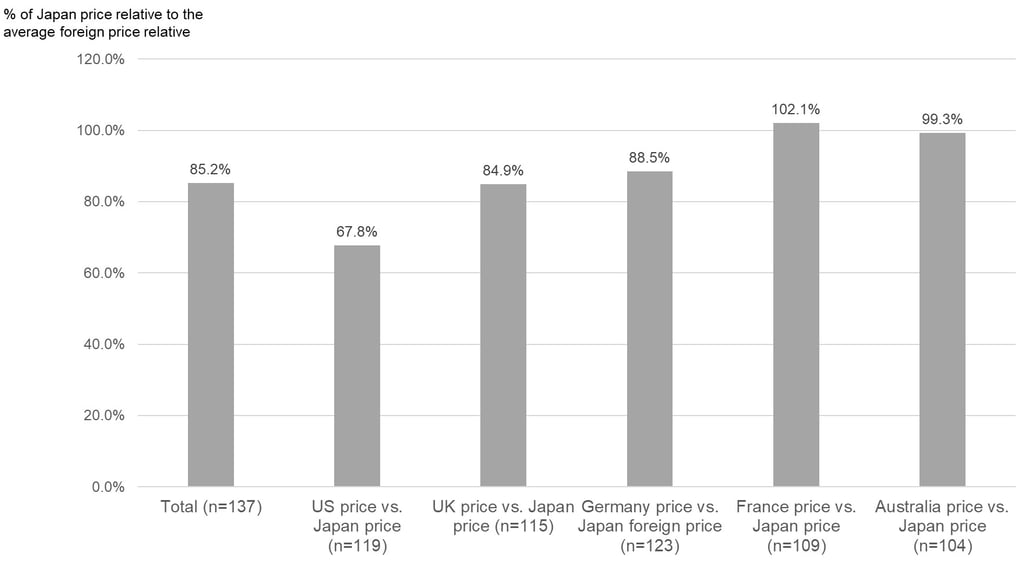
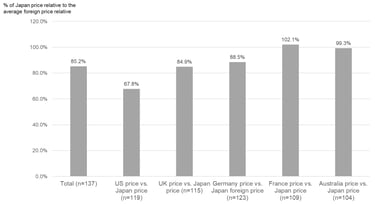
As shown in our FY 2024 new device listing here, the ratio of the initial reimbursement price for Japan to the average foreign reference price for devices newly reimbursed in FY 2024 varied significantly by device. However, the above analysis may give you an idea about the general framework for initial reimbursement prices for Japan relative to market prices in the reference markets – when one or more reference prices are available. However, you should keep in mind that foreign reference pricing doesn’t just happen at listing for devices in Japan and there is a mechanism that will adjust a new reimbursement price down by up to 50% if it exceeds the average foreign reference price by more than 1.3 times after listing. That rule, however, currently does not apply for devices (functional categories) that cover only children and rare diseases, and functional categories for which prices were revised because they were determined to be extremely difficult to supply.
Perhaps in the future we will look at how post-listing foreign reference price adjustments have affected reimbursement pricing in Japan, but we suspect that competition due to other competitor devices being listed in Japan may be a bigger driver of price reductions for devices in Japan. Nonetheless, significant reductions in device reimbursement prices outside Japan may impact prices in Japan.
For a more detailed overview or support with device reimbursement strategies in Japan, please contact us.
2025.5.17
Access Snapshot: Full-Scale 3D Heart Model Reimbursement
A submission by crossMedical, Inc. for reimbursement of its soft life-size 3D heart model was also reviewed this week, in addition to the two new device listing decisions summarized in our FY 2025 new listings page here. Their product is a life-size 3D model of the heart created based on multi-slice CT imaging information for patients with complex congenital heart disease that is difficult to diagnose and determine the surgical procedure for using existing imaging techniques. It received marketing approval in Japan on 27 July 2023 based on a clinical trial conducted in Japan.
They showed that for 20 patients with complex congenital heart disease, 65.0% (13/20: 95% confidence interval 40.8-84.6%) were not able to develop an appropriate preoperative plan without the additional information provided by their product. Moreover, in all cases the anatomical diagnosis of the product and the heart identified at the time of chest opening were consistent, and no structural incompatibility was observed. Furthermore, in 4 out of 20 cases (20%), the use of the product in preoperative planning enabled modification to the appropriate procedure. (From someone who still enjoys flipping through the Netter Atlas - this is pretty interesting technology!)
crossMedical submitted for Category C2 reimbursement in Japan, meaning that a new technical fee is also needed. They asked for 419,733 JPY per procedure based on the Cost Calculation Method. However, it was decided that it would only receive a new technical fee in Japan. Provisionally they will be allowed reimbursement based on "K939 Additional fee for surgical support such as imaging (2) By using full-scale three-dimensional models of organs" with a slightly lower technical fee of 180,000 JPY per procedure. They projected about 569 patients per year at peak annual sales after 2 years – although that was based on their requested reimbursement amount of 419,733 JPY per procedure.
This is an example of how the device reimbursement submission process in Japan sometimes does not lead to a reimbursement of the device itself (even if it is requested!), but it can trigger the establishment of a new technical fee. For more examples of devices that took that approach – either intentionally or not – please see our post below.
Please contact us if need support with device reimbursement for Japan!
2025.4.14
Technology Focus Series: Comprehensive Genomic Panel Test Market Access
In June 2019, Japan’s main reimbursement advisory committee, the Central Social Insurance Medical Council (Chuikyo), recommended the reimbursement of the first comprehensive genomic profiling (CGP) tests in Japan – Chugai’s FoundationOne® CDx Cancer Genomic Profile and Sysmex's OncoGuide NCC Oncopanel System. Since then, five other CGPs have been listed in Japan including, more recently, Otsuka’s HemeSight, which is the first gene panel test for hematological malignancies developed through a collaboration between Otsuka, Ilumina, and the National Cancer Center of Japan. HemeSight’s listing is described in the FY2024 new listings here.
The term CGP is typically reserved for gene panel tests that test for 100 or more different genes / gene mutations. The table below shows the CGPs that are currently reimbursed in Japan as well as some other reimbursed gene panel tests. All the CGP tests and other gene panel tests reimbursed to date in Japan have not received reimbursement for the test kit / system itself, but rather a new technical fee has been established for their use.
For most CGPs, hospitals receive a specific technical fee when they are used to determine treatment after standard of care (SoC) has been completed (or is expected to be completed) or when patients are ineligible for SoC. In that situation, facilities are reimbursed JPY 440,000 (approx. USD 3,064) when administering the test and then JPY 120,000 (USD 836) after an expert panel has convened and the patient is explained the results of the test. The patients would typically pay 20-30% of all costs out-of-pocket. Also, there is typically a rule that patients can only undergo a CGP once.
Most CGPs and other gene panel tests can also be reimbursed as a companion diagnostic (Dx) to determine whether a treatment can be prescribed or not, like other single gene tests. For example, among other things, Chugai’s FoundationOne® CDx CGP can be used to test for an EGFR mutation, the ALK fusion gene, or a BRAF gene mutation to prescribe Giotrif (afatinib), Zykadia (ceritinib), or Zelboraf (vemurafenib), respectively. When the test is used as a CDx, however, a lower reimbursement price applies depending on the number of items being tested for, which ranges from JPY 25,000 (approx. USD 174) to JPY 202,000 (USD 1,407). This is similar to how the reimbursement of other gene panel tests is handled. Again, the patients would typically pay 20-30% of those costs out-of-pocket. Interestingly, it seems that even if the CGP is used as a CDx to determine initial treatment, the difference in the CDx reimbursement (JPY 25,000 to JPY 202,000) and the CGP reimbursement (JPY 560,000) can be claimed by hospitals if they convene an expert panel after initial treatment has been completed and explain the test results to patients.
Exact Science’s Oncotype DX Breast Recurrence Score® Test, which is used to assess the likelihood of breast cancer recurrence in patients with early-stage, hormone receptor-positive, HER2-negative breast cancer and to determine optimal treatment, has a different fee associated with it that is slightly less than the testing fee for other CGPs. Also, the reimbursement price for GenMineTOP CGP system and HemeSight when used as a CDx was not found (It may be that kind of use is not permitted for those tests yet.)
Concerning other CGP manufacturers, it seems that Illumina’s TruSight Oncology CGP system has not been approved or reimbursed in Japan yet, but that Illumina submitted for manufacturing and marketing approval for it in May 2022. SB Tempus also recently agreed to acquire all shares of Konica Minolta Real, which markets the GenMineTop CGP in Japan. Activities of other CGP companies in Japan like Caris Life Sciences could not be confimed, but it seems like some companies such as Natera are collaborating with partners in Japan to develop evidence for personalized gene testing.
Establishment of reimbursement of gene panel testing in Japan in June 2019 was also tied to the establishment of a genome information management and usage system. Under this system, the results of gene panel tests must also be submitted to the Center for Cancer Genomics and Advanced Therapeutics (C-CAT), a division of the National Cancer Center. At present there are 13 “core” hospitals that participate in the C-CAT system that play a central role in the C-CAT – in addition to be able to convene an expert panel. Then there are also 32 “base” hospitals that can also convene expert panels and collaborate with core hospitals for research. Lastly, there are about 235 “collaborative” hospitals that are able to provide CGP testing in Japan.
According to the C-CAT site (and other MHLW materials), as of 28 February 2025, 97,965 test results had been submitted to the C-CAT system. It seems that over two-thirds of the test results submitted to date have been for Chugai’s FoundationOne® CDx CGP, but recently the percentage of submissions that comprises other tests has been increasing - including Chugai’s FoundationOne® Liquid CDx CGP. The tumor types the tests were conducted for vary a lot, but it seems like most have been submitted for pancreatic, liver, and gall bladder cancers (34%), digestive tract cancers (22%), breast cancer (7%), and lung cancer (6%). It seems, however, that only about 45% of submissions have led to a treatment option being presented by an expert panel and among those about 70% have been for the use of an approved / reimburse treatment regiment, while the remaining have been for treatment administered through a clinical trial or for compassion use, for example.
Patients who undergo cancer gene panel testing are also asked whether they agree to their data registered in C-CAT being provided to third parties for the purposes of academic research and drug development (i.e., secondary use) and it seems that nearly 100% of patients have agreed to that kind of usage which is used by the C-CAT and its affiliates for research on treatment.
Closing thoughts:
Japan was quick to establish the C-CAT to allow for secondary use of data as part of reimbursement which is no doubt a valuable resource for considering optimal treatment. In fact, it seems that about 70 research groups and around 10+ manufacturers have used the data (click here). In terms of reimbursement, it is unclear how reimbursement of CGPs in Japan “stacks up” to reimbursement in other developed countries like the US, UK, Germany, and France. However, there is a clear pathway for the reimbursement of CGP for usage after initial treatment and an option for usage as a CDx for determining initial treatment as well. Findings from Matsubara et al. (2023) suggest that “CGP testing before SoC for patients with advanced solid tumors may be clinically beneficial to guide the subsequent anticancer therapies”. Moreover Tang et al. (2023) suggest that CGP testing before SoC in Japan “may improve patient outcomes in various cancer types with a limited and controllable increase in medical costs”. So there seems to be a good case for using CGP tests earlier than later. Nevertheless, continual evidence to support that kind of use is probably needed – especially since CGPs cover a wide range of tumor types. Please contact us if you are interested in developing evidence on the benefit, utility, cost-effectiveness of CGPs and other gene panel tests for Japan.
Comprehensive Genomic Profiling (CGP) and Other Gene Panel Tests Reimbursed in Japan (as of April 2025)
*CGP reimbursement (other than OncoType DX) includes reimbursement for the test (D006-19) and the test results fee (B011-5). CDx reimbursement depends number of items being tested for in relation to the the treatment being considered.
**CRC: colorectal cancer, NSCLC: non-small lung cancer, BCa: breast cancer, PCa: prostate cancer, Mel: melanoma, TCa: thyroid cancer, CCA: (gall bladder cancer), OCa: ovarian cancer, Hem: hematologic cancers including lymphoma, leukemia, and multiple myeloma.
2025.3.21
Medical Device HTA Decisions in Japan (as of March 2025)
On March 12 the Central Social Insurance Medical Council, or Chuikyo, which is Japan's advisory group on healthcare reimbursement announced a decision on the healthcare technology assessment (HTA) of W.L. Gore & Associates' GORE TAG Conformable Thoracic Stent Graft system (hereafter, GORE TAG). This is the 3rd HTA decision for a reimbursed medical device in Japan since the launch of Japan's HTA system in April 2019. In Japan, HTA is considered after the listing of a new reimbursed medical device, drug, or regenerative medicine product and only products that meet specific criteria are selected for HTA. Hasegawa et al. (2021) provide an detailed overview the HTA process in Japan.
When products are selected for a formal HTA in Japan, the manufacturer must submit a cost-effectiveness analysis (CEA) within 9 months of the listing decision of the product or they will lose their reimbursement premium and/or face a reduction in their operating profit ratio. Please see our publication here for a detailed overview of the HTA process in Japan and other CEAs conducted for reimbursed medical devices in Japan - as well as an overview of the two previous HTA decisions for reimbursed medical devices in Japan, namely Medtronic's Micra Transcatheter Pacing System (hereafter, Micra AV) and Johnson and Johnson's Expedium Verse Fenestrated Screw System (hereafter, Expedium).
The table below provides overview of the HTA evaluation for the GORE TAG and the analysis framework used - as well as the evaluation and framework for the other two medical device HTA decisions (Micra AV and Expedium). GORE TAG was selected for HTA review in Japan based on having received a premium of 5% for its reimbursement price relative to the comparator (i.e. conventional stent grafts) due to its improvement in function and having projected peak annual sales of 8.13 billion JPY (approx. 54.4 million USD), assuming 9-year peak sales. The premium for GORE TAG was awarded because it was said to provide a safer treatment due to an improvement function - namely a lower incidence of post-operative leakage. (Note: GORE TAG is actually an example of one of a few products that was re-evaluated for reimbursement based on the "challenge submission" process introduced in Japan in April 2020, which allows devices with limited data at initial approval to be re-evaluated for reimbursement after listing. We will share more info on that system in a future update).
The manufacturer of GORE TAG conducted a cost-utility analysis (CUA) from the payer perspective as recommended by the pharmacoeconomic guidelines for Japan. A Markov model with four health states (initial surgery, postoperative follow-up, re-intervention, and death) was used. However, the analysis model assumed that the quality-of-life scores did not vary by health status, so ultimately a cost-minimization analysis (CMA) was conducted. The manufacturer analysis found a cost savings of -615,824 JPY per thoracic aortic aneurysm (TAA) case and -664,428 JPY per Stanford type B aortic dissection (TBAD) case. The public analysis group also conducted a CMA and found a cost savings of -615,824 JPY per TAA case and -138,594 JPY per TBAD case. (The reason for the lower cost savings for TBAD for the public analysis is not entirely clear, but the manufacturer assumed slightly higher follow-up costs for the comparator group than the public analysis group.) Based on these results, no change (reduction) in the reimbursement price for GORE TAG was required.
Some points raised during discussions with y the Chuikyo expert committee were as follows:
(1) Concerning the analysis framework:
- The expert committee felt that the indication for patients with traumatic thoracic aortic injury is very important due to the high need, but it was allowed to be excluded from the analysis due to the limited proportion of patients that had that indication.
(2) Concerning the additional benefit:
- The manufacturer wanted to use (1) the number of devices initially implanted, (2) the rapid pacing implementation rate, (3) overall survival rate, and (4) the reoperation rate as outcome measures for the CUA, but the expert committee said it would be inappropriate to use (1) and (2) as outcome measures because it is not clear whether they contribute to improved long-term outcomes.
- The manufacturer wanted to use a specific registry, the GREAT registry, which is a registry for the previous generation of the GORE TAG product, due to the short follow-up period for an alternative registry (the SURPASS registry). But the expert committee said that the alternative registry should be used since the previous generation of the product is a comparator technology for analysis.
Lastly, it seems that the manufacturer was hoping for a price increase due to the cost savings, but since no additional benefit was demonstrated (i.e., it was not shown to be "dominant" or substantially cost-effective) and the stent graft itself is the same product for this product and the comparative control technology an increase was not allowed. (It seems that the manufacturer appealed this decision, but Chuikyo expert committee again concluded that the results of the assessment did not meet the requirements for a price increase and that the results of the public analysis group were appropriate.)
Closing thoughts:
Although about 63 devices, drugs, and regenerative medicine products have been selected for HTA review since the system was introduced in April 2019, only 3 of those products have been devices. So the selection of reimbursed medical devices for HTA in Japan is somewhat rare. This is primarily due to the fact that most devices do not meet the peak annual sales threshold of 5 billion JPY (approximately 33.5 million USD) per year for HTA selection. However, if selected, a manufacturer must show cost-effectiveness based on a threshold of 5 million JPY (approximately 33,500 JPY) per quality-adjusted life year (QALY) or a cost-savings AND that analysis must be upheld based on a review (or re-analysis) by a public analysis group - also referred to as the Academic Technology Assessment Group (ATAG) - or face a reduction in their reimbursement price. The assessment of Micra and Expedium led to a reduction in their reimbursement price of 8.56% and 3.07%, respectively. In both of those cases, the results of the public analysis group were substantially worse that the results of of the manufacturer analysis and the public analysis group's results were prioritized (as is almost always the case!). GORE TAG was able to avoid a reduction in their reimbursement price because the public analysis groups results also showed a cost savings for the both of the indications, but it could have easily gone less smoothly. Please contact us if you would like support with the development of economic evidence for a new device in Japan, including evidence to support an HTA for Japan.
HTA Evaluations for Reimbursed Medical Devices in Japan (as of March 2025)
2025.2.28
Reimbursement of New Medical Devices Through Procedure Fees in Japan
In Japan, generally speaking, only devices that are used for a single patient receive a separate reimbursement. Those are often referred to as special treatment materials (STM). Matsumoto et al. (2022) provide a detailed overview the reimbursement process for STMs in Japan and LoPresti et al. (2024) provide an overview of reimbursement outcomes for STMs between fiscal year (FY) 2015 and FY 2023. But, many medical devices, including recent devices that incorporate artificial intelligence for diagnostics, for example, can be used across multiple patients. Those devices typically do not receive a separate reimbursement in Japan, but rather the cost for using them must be covered through procedure (or technical) fees which are set at the national level.
Some devices may also be broken out so that the disposable (consumable) part of the device receives a separate reimbursement, but the non-disposable part of the device, which can be used for multiple patients, is reimbursed through a procedure fee. This can advantageous because procedure fees tend to be less susceptible to biennial reimbursement revisions (reductions). If the procedure fee is sufficient enough to cover the purchase of the device in the long-run, then a clinic or hospital may have an incentive to purchase and use of the device for their patients. On the other hand, if the procedure fee is insufficient to allow a clinic or hospital to achieve an adequate return on their investment, then they may have less incentive to purchase and use it.
There are basically two approaches to establishing a procedure fee in Japan: (1) a request submitted by (through) a medical society or (2) submission through the STM reimbursement process. The Central Social Insurance Medical Council, or Chuikyo, which is Japan's advisory group on healthcare reimbursement, reviews about 700-750 new or revised procedure (technical) fee requests from medical societies every 2 years in Japan and about 15-20% of those requests are approved. A more direct approach to establish a new procedure fee is to submit for reimbursement of the device as an STM in order to trigger the establishment of a new procedure fee for the device. While new or revised procedure fees are only introduced every 2 years, submission through the STM reimbursement process can allow for the use of the device based on a preliminary procedure fee by referencing an existing procedure fee to be used before a new procedure fee can be established. This allows clinics and hospitals (and ultimately patients) to receive a procedure fee when the device is used in the interim until a new procedure fee is formally established.
Among the roughly 215 unique new devices assessed for reimbursement in Japan between FY 2015 and FY 2024, about 70 devices were not reimbursed based on a separate reimbursement, but rather they were evaluated for a new procedure fee. The table below shows some example of those products, the reimbursement they requested, and the preliminary procedure fee they were allowed at launch. As shown in the table, for many devices submitted through the STM reimbursement process that were reimbursed based on a procedure fee, the manufacturer had requested that a specific procedure fee be used as the preliminary procedure fee before a new procedure fee is established. In most cases, however, the preliminary procedure fee received was lower than the fee requested.
On the other hand, many devices had requested a separate reimbursement, but did not receive it. Instead, they received a reimbursement based on a procedure fee, so if the procedure is not conducted then there is no reimbursement for the use of the device. It is not clear in those cases whether the manufacturer knew they had a strong chance of being denied a separate reimbursement, but still took a chance on getting a separate reimbursement - or if they actually thought they might be able to get a separate reimbursement for the product.
Closing thoughts:
Working with medical societies to establish a new (or revised) procedure fee can be time consuming and the probability of success is not particularly high. As such, submission for reimbursement as an STM is an important option for establishing a procedure fee for a new device that is not a single-use device. This is particularly important if the new device includes both components that are single-use and those that are not. As such, it is important to understand how the STM reimbursement process works in Japan and to plan ahead. GinePro can support with strategy development and planning and can help facilitate the reimbursement of novel medical devices in Japan. Please contact us for more information.
Examples of Medical Devices Reimbursed Through Procedure (Technical) Fees (FY2015 - FY2024)
2025.1.28
Technology Focus Series: Occluder Device Market Access [Functional Categories: 131, 153, 204, and 205]
Reimbursement of Occluder Devices in Japan [Functional Categories: 131, 153, 204, and 205]
2025.1.14
Technology Focus Series: Guiding Catheter Market Access [Functional Category 132]
In this Technology Focus Series post, we will discuss market access issues for guiding catheters which are reimbursed in Japan based on the “132 Guiding catheters” functional category grouping. Let’s begin with a brief overview of guiding catheters, for those with less experience in the area. A guiding catheter is a long, flexible, and hollow tube (shaft) that can be inserted into blood vessels to allow access for other, narrower catheters, for example. They have a proximal end, which is handled by the physician, and a distal end / tip that is designed for optimal placement and control near a lesion or area in the body. They are often made from materials like polyurethane (PU) or polyethylene (PE), which allows for flexibility and pushability - i.e., the ability to transmit force between the proximal end and the distal end / tip. In some cases, more than one guiding catheter may be used per patient / procedure.
As their name implies, guiding catheters are used to help navigate through the vascular system to reach specific areas like the heart, brain, kidney, limbs, etc. and to provide support and stability when delivering other devices such as balloons, stents, and ablation catheters that are used to treat lesions, calcifications, thrombi, etc. in the blood vessels. In that sense, guiding catheters are an essential tool for many interventional (minimally invasive) procedures and have reduced the need to rely on surgical intervention.
Note: There is another device called a guidewire which can serve a similar function as guiding catheters. However, guidewires differ in their design and function from guiding catheters. As an analogy, if guiding catheters are the transport vehicles or armored personnel carriers (APCs) for the staging of interventional procedures, then guidewires are reconnaissance units that also support with initial transport tasks for the assault teams (e.g., balloons and stents). Similar to guiding catheters, guidewires are long, thin devices that differ in their diameter and length, as well as stiffness. They act as a rail for the insertion of catheters, including guiding catheters, and different guidewires may be used for different types of blood vessels and locations. We may cover guidewires in a future Technology Focus Series post, but they tend to be spread out across different functional category groups as opposed to having a stand-alone group like “132 Guiding catheters”.
Below is a table showing the different functional categories included in the “132 Guiding catheter” group including information on when they were established, examples of products they include, examples of relevant procedures, their approximate volume in fiscal year (FY) 2022, and their initial and current reimbursement in Japan (as of FY 2024). The two oldest functional categories in this group are “132 (1) For coronary arteries” and “132 (2)-1 For cerebral blood vessels: standard type” which were both established in FY 2002. Guiding catheters reimbursed as “132 (1) For coronary arteries” include products like Medtronic’s Sherpa NX, Terumo’s Heartrail II, and Boston Scientific’s Mach I, for example. Those reimbursed as “132 (2)-1 For cerebral blood vessels: standard type” include products used to deliver endovascular microcatheters, for example, safely to the surgical site of the cerebral blood vessel when performing cerebrovascular surgery. This includes products like Medikit’s Axcelguide, Asahi Intec’s FUBUKI XF, and Terumo’s VIA series, for example.
Initially the market for guiding catheters for coronary arteries was dominated by multinational manufacturers like J&J (Cordis), Medtronic, and Boston Scientific. But, in recent decades, it seems that domestic manufacturers like Terumo and Asahi Intec have gained a lot of market share. The market for guiding catheters for the cerebral (and peripheral) blood vessels, on the other hand, has always had several strong domestic players. However, both of these two oldest functional categories in this group now have 10+ manufacturers for Japan. Moreover, their reimbursement price has fallen substantially since initially established with a 78% decline for the “132 (1) For coronary arteries” functional category and 30% decline for “132 (2)-1 For cerebral blood vessels: standard type” functional category.
The second largest functional category for guiding catheters in FY 2022 was the “132 (3) For other blood vessels” functional category, which was introduced in 2018. Similar to the “132 (2)-1 For cerebral blood vessels: standard type” functional category, this category includes products used to the deliver endovascular microcatheters safely to the surgical site of the blood vessel when performing procedures for peripheral blood vessels – particularly those other than coronary artery and cerebral blood vessels. Examples of those products include Medikit’s Parent series, Goodman’s Profit series, and Cook Medical’s FLEXOR SHUTTLE / ANSEL product. For this functional category there are also 10+ manufacturers in Japan. Moreover, the reimbursement price has declined by 10% since it was established.
More recently established functional categories in this group include the “132 (2)-4 For cerebral blood vessels: radial artery puncture compatible” functional category and the “132 (4) For bronchial tube” which were both established in FY 2024. Both appear to have only one product / manufacturer at the moment, namely the Rist™ radial access system and the Chartis Pulmonary Function Assessment system, respectively. The former requested reimbursement with the “132 (2)-2 For cerebral blood vessels: highly-flexible type” functional category used as the baseline comparator and a 10% Improvement premium, which would have allowed for a reimbursement price of 96,200 JPY. But they were instead reimbursed using the cost-based approach and ended up with a reimbursement of 63,200 JPY and no premiums. They had shown a substantially lower rate of bleeding at the puncture site (0.3% vs. 5.57%) compared to the standard transfemoral access (TFA) approach.
The Chartis Pulmonary Function Assessment system was listed together with Pulmonx’s Zephyr for COPD patients with severe emphysema. Prime Fine had requested reimbursement with the “169 (2) Material with pressure sensor for angiography: Catheter with pressure sensor for angiography” functional category used as the baseline and a 10% Usefulness premium, which would have allowed for a reimbursement price of 140,000 JPY. But they were instead reimbursed in comparison with the “132 (2)-2 For cerebral blood vessels: high flexible type” functional category and ended up with a reimbursement of 90,300 JPY and no premiums. (Note: Asking for a higher reimbursement but ending up with something less is very common in Japan.)
Closing thoughts:
A substantial decline in reimbursement over time is not uncommon for reimbursed medical devices in Japan. There are thought to be several drivers for this including greater competition and foreign reference pricing, for example. The reimbursement price of reimbursed medical devices is reviewed at least every two years in Japan based on the results of a wholesaler and provider survey to consider the gap between the market price and the reimbursement price. To the extent that manufacturers discount their products to hospitals due to competition, for example, they may face a reduction in their reimbursement price. Moreover, unlike pharmaceutical products, reimbursement prices for most reimbursed medical devices are reviewed relative to the foreign reference price (US, UK, Germany, France, and Australia) at least every two years – not just at initial listing - and reduction may apply if the price in Japan is higher by more than 1.3 times the average foreign reference price taking into consideration local consumption taxes.
Another notable aspect of the reimbursement of guiding catheters in Japan, and indeed many other medical devices in Japan, is that even though a similar technology may be used, the reimbursement price in Japan may differ depending on the purpose and area that the device is used for. This appears to be the case for Goodman’s Profit series, for example, which has products for both the “132 (1) For coronary arteries” functional category and “132 (2)-1 For cerebral blood vessels: standard type” functional category, which have very different reimbursement prices (8,220 JPY vs. 21,800 JPY). But there are other examples as well. This raises the question about whether these devices (and many others) are priced according to their value. On the other hand, these product after have subtle differences in terms of design and materials used, so it could be argued that while the once-fits-all, functional category approach to reimbursement may be simpler, it may not be appropriate for areas where material and manufacturing costs can be vary greatly by the specific design (e.g., length / diameter, etc.).
Please contact GinePro if you would like a detailed description of the reimbursement for a specific medical device area in Japan.
Reimbursement of Guiding Catheters in Japan [Functional Category 132]
2024.12.23
Examples of the New Economic Efficiency Premium
In April 2024, a new reimbursement premium was introduced in Japan for reimbursed medical devices, the Economic Efficiency premium. While there are several premiums available for newly reimbursed devices in Japan which are described in detail here, this is a reimbursement premium unlike any that we have seen before in Japan. This premium allows for part of the cost savings from the use of a new device to be incorporated directly into the reimbursement price for the new device. This premium is applied when a new medical device (1) targets essentially the same condition and has the same intended use as an already listed device, (2) it demonstrates clinical efficacy that is equal to or better than the already listed device, (3) it can serve as a substitute for the already listed device, and (4) it is expected to reduce costs related to insurance medical devices compared to using the already listed device. With this premium, 50% of the anticipated cost savings from eliminating the use of other devices is awarded as a premium for the per unit reimbursement price of the new medical device.
This new premium was introduced based on recommendations from the device industry. The basic principle if that even if a new device is more expensive than existing devices, it may lead to a reduction in costs overall if it allows for the elimination of use of other devices. The example given in the reimbursement revision report is the case whereby, while conventional deep brain stimulation (DBS) therapy for Parkinson’s disease might require two devices with two leads (e.g., Medtronic Activa PC/S/RC), a new device might allow for just one device with two leads (e.g., Medtronic Percept PC). With the new premium, 50% of costs savings from eliminating the need for other devices may be incorporated into the reimbursement of the new device.
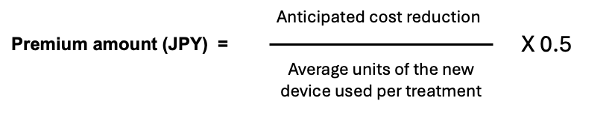
The premium amount itself is calculated by dividing the anticipated cost reduction with the new device by the anticipated average number of units of the device used per treatment and multiplying that amount by 0.5 as show below:
The cost reduction is “said to be” calculated by subtracting the product of the average number of units of the new device and the reimbursement price of the already listed device from the product of the average number of units of the already listed device and reimbursement price of the already listed device. This sounds rather like a very basic budget impact analysis, which all devices must include in their dossier submission. But, looking at a recent example of its application, the calculation for the premium seems a bit more straight-forward and is more in line with the intention of the premium as described in the 2nd paragraph above.
Since April 2024, we have seen this premium awarded for two newly listed devices: Johnson & Johnson’s VARIPULSE Pulsed Field Ablation Catheter and Medtronic’s PulseSelect PFA Loop Catheter. Both were listed in September 2024 and a detailed overview of their reimbursement decision can be found here. For both devices the projected cost savings was determined by using the “114 Catheter electrode for extracorporeal pacemakers: (2) Cardiac electrophysiology test function added type 2) Coronary sinus type" functional category which is 64,000 JPY per unit. As such, the premium allowed was 32,000 JPY per unit (64,000 JPY x 0.5 = 32,000 JPY). So, the notion seems to be that when using the VARIPULSE Pulsed Field Ablation Catheter or the PulseSelect PFA loop, the use of a coronary sinus-type catheter electrode for extracorporeal pacemakers that has cardiac electrophysiology test functionality (e.g., Johnson & Johnson’s Lasso NAV 20mm, etc.) can be eliminated.
It is hoped this new premium will help mitigate any disincentives for manufacturers to create more economic efficient versions of existing technologies, whereby launches those products might mean that products that they already sell may be used less or become obsolete. Since it can be challenging for medical devices to prepare cost-effectiveness analysis prior to listing due to lack of data incentives that reward technological innovation like this new premium are very encouraging and will help to improve access to new technologies for patients in Japan.
Closing thoughts:
Our feeling is that the Economic Efficiency premium is a big step for medical device reimbursement in Japan – which is an area where more incentives for introducing new technologies are really needed. It is not uncommon for new premiums (or rationale for premiums) to be established for medical devices without much practical usage or application. (Please see our publication here for more information on that problem.) So, it is encouraging that we have already seen some examples of this premium being applied. However, manufacturers must be prepared to present evidence on how their new device will affect the use of other devices when reimbursement is considered. As mentioned above, a basic budget impact analysis is required for all new devices (and procedures) in Japan. So, a simple supplemental analysis that shows how the use of other devices may be reduced could be done in addition to that analysis. GinePro has expertise with conducting budget impact analyses for new medical devices in Japan and we can support with an assessment of a potential Economic Efficiency premium as part of that process. Please contact GinePro if you would like to hear more about those services.
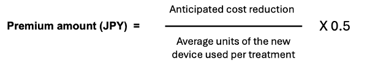
2024.12.9
Technology Focus Series: Regenerative Medicine & Cellular Therapy Market Access in Japan
In 2014, Japan implemented two new laws that effectively established regenerative medicines and cellular therapies as a separate regulatory category in Japan - the Act on the Safety of Regenerative Medicine and the Pharmaceuticals, Medical Devices, and other Therapeutic Products Act. More specifically, the following kinds of products (A or B) are reviewed by the regulatory bodies in Japan as regenerative medicines or cellular therapies:
A. Products that involve processes such as the culturing of human or animal cells and that (1) allow for the regeneration, restoration, and/or reformation of the structure or functionality of the body and (2) are meant to be used for prevention or treatment of a condition
B. Products that introduce human cells to the body as gene therapy
As of December 2024, at least 20 regenerative medicine products had been approved for marketing and 19 had been listed for reimbursement in Japan. Moreover, nearly 20 other regenerative medicine and cellular therapy products have received orphan designation and/or Sakigake (breakthrough) designation - meaning that they have a relatively high chance of development success and receiving regulatory approval in the not-too-distant future.
The reimbursement of regenerative medicines in Japan is determined based on whether the product is considered closer to a drug agent or a medical device. JCR’s Temcell HS mesenchymal stem cells and Novartis’s Kymriah (tisagenlecleucel), for example, were reimbursed using the drug reimbursement process. However, Terumo's HeartSheet autologous skeletal myoblast-derived cell sheet and, more recently, Aurion Biotech’s Vyznova (nelependocel), for example, were reimbursed as a medical device. The table below provides a list of the regenerative medicine and cellular therapies that were reimbursed in Japan as of December 2024 – as well as the approach to reimbursement that was used and their reimbursement outcome.
Out of the 20 regenerative medicine and cellular therapy products that are currently listed, 9 products were reimbursed using the medical device reimbursement pathway. That typically includes products that are cellular therapies applied to the body like J-TEC’s autologous epitheleal and joint treamtents (JACE, JACC, Nepic, etc.) and, more recently, Aurion Biotech’s in vitro corneal endothelial cells treatment, Vyznova (nelependocel), for bullous keratopathy. Note that as of December 2024, Janssen’s Carvykti (ciltacabtagene autoleucel) and SanBio’s Akuugo (vandefitemcel) have also received marketing approval in Japan, but have not yet been listed for reimbursement.
It is also common for regenerative medicine and cellular therapies to be reimbursed using a cost-based approach – likely because they tend to be quite novel in terms of their mechanism of action and molecular structure. On the other hand, in the case of CAR T treatments, the first CAR T therapy, Kymriah (tisagenlecleucel), was reimbursed using the cost-based approach, but subsequent CAR T therapies (Yescarta, Breyanzi, Abecma) have all been reimbursed in comparison to Kymriah with no premiums allowed – even though some of those newer treatments have shown different outcomes and/or target different conditions. This issue has, in part, driven some discussion about the establishment of a separate reimbursement process for regenerative medicines and cellular therapies which is apparently still underway.
Several products had received orphan designation and/or Sakigake (breakthrough) designation. Please see the updates below for an overview of those designation and their potential impact on reimbursement.
Two products, Terumo’s HeartSheet and AnGes’s Collategene, have recently discontinued sales due to inability to meet requirements of their conditional approval (i.e., inability to shown efficacy based on post-marketing surveillance). Two other products, Nipro’s Stemirac autologous mesenchymal stem cells and Daiicihi-Sankyo’s Delytact (teserpaturev), also received conditional approval and will have to submit for formal approval when their review period are completed. Both products received a review period of 7 years, so by our calculations Stemirac’s should be up for review later this year and Delytact will be up for review in 2028. SanBio’s Akugo (vandefitemcel) also received conditional approval earlier this year, but it has yet to be listed in Japan.
Another important point is that Optimal Clinical Use Guidelines (hereafter “optimal use guidelines”) have been established for some regenerative medicine and cellular therapy products – namely all of the CAR T therapies (Kymriah, Yescarta, Breyanzi, and Abecma), Stemirac, and Delytact (teserpaturev). These guidelines are developed by medical societies in Japan at the request of the reimbursement authorities, and they further limit the reimbursement of products by specifying the kinds of patients that can receive them (in even more detail than label itself) and the hospital settings that they can be administered in. Optimal use guidelines are issued to ensure that innovative drugs and regenerative medicines are provided to the most appropriate patients. Elsewhere it has been stated that they are meant to help address the increasing number of expensive drugs in Japan. Notably, however, optimal use guidelines have not been requested for many other regenerative medicine and cellular therapy products, some of which are even more expensive those mentioned above, including Zolgensma (onasemnogene abeparvovec), which is currently Japan’s most expensive treatment at 167,077,222 JPY (or about 1.1 million USD) per administration. This may be because those products do not involve as detailed of a delivery process or that their risk of over-usage and/or extended indications is not as high. Nevertheless, new regenerative medicine and cellular therapy products could be asked to prepare optimal use guidelines in the future at the time of listing.
Closing thoughts:
Since Dr. Shinya Yamanake received the Nobel Prize for his work on induced pluripotent stem (iPS) cells in 2021, Japan has been a leader in the development of regenerative medicine and cellular therapy products. Moreover, to our knowledge, Japan is one of the first countries to establish a somewhat transparent reimbursement pathway for those products. However, the ability for Japan to reimburse novel new regenerative medicine and cellular therapy products may come into crisis as the availability of those treatments increase. The listing of new gene therapies to treat hemophilia conditions such as Biomarin’s valoctocogene roxaparvovec, CSL Behring’s etranacogene dezaparvovec, and Pfizer giroctocogene fitelparvovec, will probably “stir the coals” of that debate further over the next couple of years. In other countries, novel risk-sharing agreements such as pay-for-performance agreements and capitation-based agreements have become more prevelanet with the emergence of regenerative medicine and cellular therapy products. However, those kinds of agreements currently do not exist in Japan. This is primarily due to the fact that current reimbursement system in Japan does not really allow for retrospective settling of payments between patients, insurers, and providers. Another consideration, however, is HTA review. One of the key selection criteria for an HTA review in Japan is that the new treatment has “a remarkably high reimbursement price”. While the formal threshold for “a remarkably high reimbursement price” has not been defined, many regenerative medicine and cellular therapy products are likely to meet this criterion. Products selectedf for HTA review are asked to submit a cost-effectiveness analysis (CEA) following intiial listing and their reimbursement may be reduced by a small, but non-trivial amount if they are unable to show cost-effectiveness. Since the start of the HTA system in Japan in April 2019, all the CAR T treatments have been selected for HTA review as well as Zolgensma (onasemnogene abeparvovec). The CAR T review led to about a 4% reduction in the reimbursement rate of those products. However, the review for Zolgensma has been postponed until more data can be collected to allow for a CEA to be conducted. Regardless, it is important to think ahead concernig the potential challenges of obtaining and defending the approval and reimbursement status of regenerative medicine and cellular therapy products in Japan through well-designed and well-managed post-marketing surveillance studies and health economic studies following listing. GinePro can assist with these aspects, if needed, so please drop us a line if you need support.
New Regenerative Medicine and Cellular Therapy Product Approvals and Listings for Japan (As of December 2024)
2024.11.11
Breakthrough Designation for Devices and Regenerative Medicine Products in Japan
In 2015, Japan introduced a new breakthrough designation for treatments in Japan. That system is referred to as the Sakigake designation system and products that receive that designation can benefit from an expedited regulatory review process and additional support similar to orphan designation. The key criteria for Sakigake (breakthrough) designation are as follows:
(1) the new treatment is innovative with a new mechanism of action
(2) the new treatment addresses a severe condition
(3) the new treatment is considered to be highly efficacious and/or safe
(4) the new treatment is likely to be developed prior to (or within 3 months of) other similar countries
Since April 2015, 14 device products, 12 regenerative medicine products, and 2 in-vitro diagnostic (IVD) products have received Sakigake (breakthrough) designation. For devices, more recently, Sound Wave Innovation’s IPUS-Brain transcranial low-intensity pulsed ultrasound therapy device (LIPUS-Brain) received Sakigake (breakthrough) designation for the treatment of patients with early Alzheimer’s disease in September 2024 and iCorNet Laboratory’s cardiac shape correcting net product received Sakigake (breakthrough) designation in June 2020 for reduction of excessive left ventricular wall tension and prevention of the development of cardiac remodeling in patients with non-ischemic dilated cardiomyopathy with progressive cardiac enlargement.
For regenerative medicine products, more recently, JUNTEN BIO’s inducible inhibitory T-cells (JB-101) received Sakigake (breakthrough) designation in June 2020 for the reduction or interruption of immunosuppressive drugs after liver transplantation. Moreover, SanBio’s Akuugo (vandefitemcel) suspension for intracranial implantation received Sakigake (breakthrough) designation in April 2019 as a treatment for improving chronic motor paralysis from traumatic brain injury (TBI). For IVDs, Toray Industries’s DNA chip microarray pancreatic and biliary tract cancer test kit received Sakigake (breakthrough) designation in April 2019 for the diagnosis of pancreatic and biliary tract cancer and Sysmex’s OncoGuide NCC Oncopanel system received designation in February 2019 as a comprehensive genomic profiling test for tumor tissue in patients with solid tumors.
In April 2022, a new Sakigake (breakthrough) premium was also introduced for newly listed devices that allows for a reimbursement premium of +10% if Sakigake (breakthrough) designation is received prior to listing. Since then, among the devices that received Sakigake (breakthrough) designation and were approved/listed in Japan, only one device, Teijin Medical Technologies’s novel surgical patch, SYNFOLIUM, has received a Sakigake (breakthrough) premium. Among the 3 regenerative medicine products that received Sakigake (breakthrough) designation and are already approved / listed in Japan, all 3 products received a Sakigake (breakthrough) reimbursement premium of +10%. Those products include Novartis’s Zolgensma (onasemnogene abeparvovec), Daiichi-Sankyo’s Delytact (teserpaturev), and Nipro’s Stemirac autologous mesenchymal stem cells. Lastly, for the two IVDs mentioned that received Sakigake (breakthrough) designation, the OncoGuide NCC Oncopanel system was ultimately not reimbursed as a stand-alone product and therefore did not receive a premium and information on Toray’s DNA chip microarray kit was not found.
Closing thoughts:
While Sakigake (breakthrough) designation can offer a lot of advantages when it comes to regulatory approval and potentially an additional reimbursement premium, launching in Japan early (or around the same time as other markets) is a prerequisite, so early planning is needed. In fact, a quick glance at the list(s) of device and regenerative medicine products that have received Sakigake (breakthrough) designation shows that, unlike products that received orphan designation, nearly all the products that have received Sakigake (breakthrough) designation are manufactured by companies headquartered in Japan. This suggests that early planning is needed to coordinate the development schedule in Japan alongside other markets and to obtain designation early. If you want to take advantage of this designation for your product in Japan, then please contact GinePro as early as possible so that we can help accelerate your evidence development for Japan.
2024.10.28
Orphan Designation for Devices and Regenerative Medicine Products in Japan
New medical device and regenerative medicine products that are meant to treat conditions with only a small number of patients in Japan may be eligible for orphan device or orphan regenerative medicine designation. This designation allows for assistance with development of a study design, expedited regulatory review, lower regulatory fees, a longer period of market exclusivity, and, potentially, a reimbursement premium known as the Marketability I premium.
The key criteria for orphan device designation are as follows:
(1) the new treatment targets a condition with less than 50,000 patients in Japan*
(2) the new treatment addresses a high medical need – meaning there is no adequate alternative treatment for the condition in terms of efficacy and safety
(3) there is a high probability of success for its development in Japan
Since 2015, 8 devices and 27 regenerative medicine products received orphan designation in Japan. More recently, for devices, in December 2024 Sumitomo Heavy Industries’s boron neutron capture therapy (BNCT) NeuCure treatment and dose calculation system received orphan device designation for the treatment of unresectable locally advanced or locally recurrent head and neck cancer and recurrent malignant glioma. Moreover, in March 2024 Boston Scientific’s Visual-ICE Cryoablation System received orphan device designation for renal angiomyolipoma associated with tuberous sclerosis.
Among the 8 devices that received orphan device designation since 2015, 7 devices have since been approved and listed in Japan and 2 devices received a Marketability premium at listing based on having received orphan device designation. The Marketability premium based on orphan device designation typically allows for a +10% reimbursement premium. The 2 device products that received a Marketability premium based on orphan device designation are listed below together with the peak annual patient volume that their reimbursement submissions indicated
Medtronic’s Harmony™ Transcatheter Pulmonary Valve system for patients with severe pulmonary regurgitation who have a history of surgical repair (patch repair) or transcatheter intervention (balloon valvuloplasty) to the right ventricular outflow tract and who have a clinical need for pulmonary artery valve replacement
Peak annual patients (10 years): 85 patients
Mallinckrodt’s Cellex extracorporeal photopheresis (ECP) system the treatment of steroid-resistant or intolerant chronic graft-versus-host disease
Peak annual patients (10 years): 38 patients
Some other devices that had received orphan device designation and were listed prior to April 2020 did not receive a Marketability premium, but instead received an increase in their operating profit ratio.**
Having received orphan device designation appears to also be associated with the receipt of a utility reimbursement premium (i.e., the Usefulness or Improvement premiums). Since 2015, among the 7 newly reimbursed medical devices that had received orphan device designation, 5 products also received a utility premium. More information on the utility reimbursement premium and factors associated with it can be found in our publication here.
For regenerative medicine products, more recently, Ultragenyx’s pariglasgene brecaparvovec gene therapy received orphan regenerative medicine designation in Japan in November 2024 for the treatment of glycogen storage disease type Ia (GSDIa) and in August 2024 Biomarin’s valoctocogene roxaparvovec gene therapy received orphan regenerative medicine designation for the treatment of congenital hemophilia A. Moreover, in March 2024, CSL Behring’s etranacogene dezaparvovec gene therapy received orphan regenerative medicine designation in Japan for the treatment of hemophilia B. At present there are about 20 regenerative medicine and cellular therapy products that have been approved and listed in Japan and among them, several have received a Marketability premium of +10% based on having received orphan designation. Some examples include Novartis’s Kymriah (tisagenlecleucel) for CD19-positive B-cell acute lymphoblastic leukemia, diffuse large B-cell lymphoma (DLBCL), and follicular lymphoma, which was listed in May 2019, and Takeda’s Alofisel (darvadstrocel) for the treatment of complicated hemorrhoidal fistulas in patients with inactive or mildly active Crohn's disease, which was listed in November 2021. More recently, Aurion Biotech’s Vyznova, which was listed for the treatment of bullous keratopathy in September 2024, also received a Marketability I premium based on having received orphan regenerative medicine designation.
Similar rules apply for in-vitro diagnostic (IVD) products. However, for IVDs a higher reimbursement premium is allowed for products with fewer patients (usage frequency) per year as follows:
+20%: 600 to less than 800 patients (usage frequency) per year
+30%: 400 to less than 600 patients (usage frequency) per year
+40%: 200 to less than 400 patients (usage frequency) per year
+50%: Less than 200 patients (usage frequency) per year
Moreover, for IVDs that do not receive orphan device designation but are expected be used for fewer than 1000 patients (usage frequency) per year, a +10% premium may be possible with a higher premium for products with even fewer patients (usage frequency) per year, similar to the levels shown above. Two IVDs, Nihon Stery’s AmoyDx FGFR2 Break-apart FISH Probe kit and Kohjin Bio’s KBM Lifetech aPAP kit, recently received a +20% and +50% reimbursement premium, respectively, for targeting conditions with very few patients in Japan. You can find more information on those decisions here.
*In principle, calculating the number of patients for a specific condition as less than 50,000 patients by adding criteria such as “severe patients” without a clear medical reason, is not accepted. But some exceptions are allowed when the treatment targets a condition for which there is a high unmet need and the number of patients for the condition, as a whole, is not significantly higher than 50,000 patients.
**Prior to April 2020, the reimbursement premium system only applied to newly reimbursed devices that were reimbursed using a comparative approach, and did not apply for devices reimbursed using a cost-based approach. However, devices reimbursed using a cost-based approach could benefit from an increase in their operating profit ratio if they had received orphan device designation and/or they were for conditions with very few patients.
Closing thoughts:
While there is a detailed application process for orphan device and orphan regenerative medicine designation, obtaining orphan designation may be worthwhile for devices and regenerative medicine products that target a smaller number of patients in Japan as it can lead to a higher reimbursement price and/or the price maintenance premium. If no reliable evidence is available on the number of patients in Japan, then an epidemiology study using real-world data for Japan, for example, can be used to support the number of patients. But the evidence should be published in a peer-reviewed journal prior to finalization of the orphan designation submission. Moreover, the number patients reported to the authorities in Japan (e.g., the PMDA and MHLW) should be consistent throughout the development process to avoid misunderstandings and a reduction in the credibility of the evidence presented. GinePro can advise and support you with developing the evidence needed for this process, so please contact us if you need support.
We respect your privacy. You will NOT be sent relentless emails by GinePro!


Healthcare evidence generation & access support for Japan.
© 2024-2025 GinePro LLC. All rights reserved.